enrichR module¶
- class magine.enrichment.enrichr.Enrichr(verbose=False)[source]¶
Bases:
object- run(list_of_genes, gene_set_lib='GO_Biological_Process_2017')[source]¶
- Parameters
- list_of_geneslist_like
List of genes using HGNC gene names
- gene_set_libstr or list
Name of gene set library To print options use Enrichr.print_valid_libs
- Returns
- dfEnrichmentResult
Results from enrichR
Examples
>>> import pandas as pd >>> pd.set_option('display.max_colwidth', 40) >>> pd.set_option('precision', 3) >>> e = Enrichr() >>> df = e.run(['BAX', 'BCL2', 'CASP3', 'CASP8'], gene_set_lib='Reactome_2016') >>> print(df[['term_name','combined_score']].head(5)) term_name combined_score 0 intrinsic pathway for apoptosis hsa ... 11814.410 1 apoptosis hsa r-hsa-109581 2365.141 2 programmed cell death hsa r-hsa-5357801 2313.527 3 caspase-mediated cleavage of cytoske... 10944.261 4 caspase activation via extrinsic apo... 4245.542
- run_samples(sample_lists, sample_ids, gene_set_lib='GO_Biological_Process_2017', save_name=None, create_html=False, out_dir=None, run_parallel=False, exp_data=None, pivot=False)[source]¶
Run enrichment analysis on a list of samples.
- Parameters
- sample_listslist_like
List of lists of genes for enrichment analysis
- sample_idslist
list of ids for the provided sample list
- gene_set_libstr, list
Type of gene set, refer to Enrichr.print_valid_libs
- save_namestr, optional
if provided it will save a file as a pivoted table with the term_ids vs sample_ids
- create_htmlbool
Creates html of output with plots of species across sample
- out_dirstr
If create_html, it will place all html plots into this directory
- run_parallelbool
If create_html, it will create plots using multiprocessing
- exp_datamagine.data.ExperimentalData
Must be provided if create_html=True
- pivotbool
- Returns
- EnrichmentResult
Examples
>>> import pandas as pd >>> import matplotlib.pyplot as plt >>> from magine.enrichment.enrichr import Enrichr >>> pd.set_option('display.max_colwidth', 40) >>> pd.set_option('precision', 3) >>> samples = [['BAX', 'BCL2', 'CASP3', 'CASP8'], ['ATR', 'ATM', 'TP53', 'CHEK1']] >>> sample_ids = ['apoptosis', 'dna_repair'] >>> e = Enrichr() >>> df = e.run_samples(samples, sample_ids, gene_set_lib='Reactome_2016') >>> print(df[['term_name','combined_score']].head(5)) term_name combined_score 0 intrinsic pathway for apoptosis hsa ... 11814.410 1 apoptosis hsa r-hsa-109581 2365.141 2 programmed cell death hsa r-hsa-5357801 2313.527 3 caspase-mediated cleavage of cytoske... 10944.261 4 caspase activation via extrinsic apo... 4245.542 >>> df.filter_multi(rank=10, inplace=True) >>> df['term_name'] = df['term_name'].str.split('_').str.get(0) >>> fig = df.sig.heatmap(figsize=(6, 6), linewidths=.05)
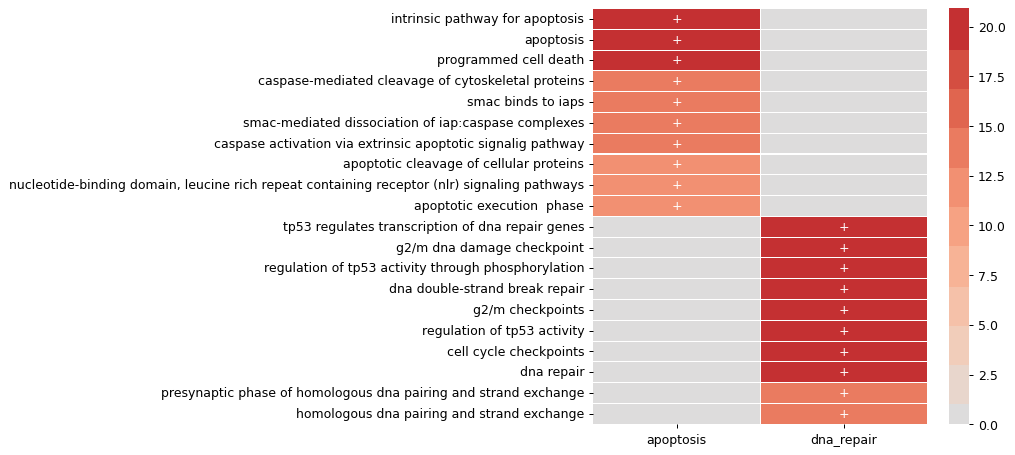
Using a ExperimentalData instance, we can run enrichR for all the databases using a simple wrapper around.
- magine.enrichment.enrichr.run_enrichment_for_project(exp_data, project_name, databases=None, output_path=None)[source]¶
- Parameters
- exp_datamagine.data.experimental_data.ExprerimentalData
- project_namestr
- databaseslist
- output_pathstr
Location to save all individual enrichment output files created.
We also provide some tools to clean up and standardize enrichRs output.
Functions to cleanup enrichR term names¶
Note: this are in progress and not fully tested! Warning!